Das Klippel-Trénaunay-Syndrom (KTS, auch Klippel-Trénaunay-Weber-Syndrom oder auch angiektatischer Riesenwuchs) ist eine angeborenes Fehlbildungssyndrom. Es tritt nur sporadisch auftritt, wird also nicht vererbt. In der überwiegenden Zahl der Fälle ist nur eine Extremität betroffen.
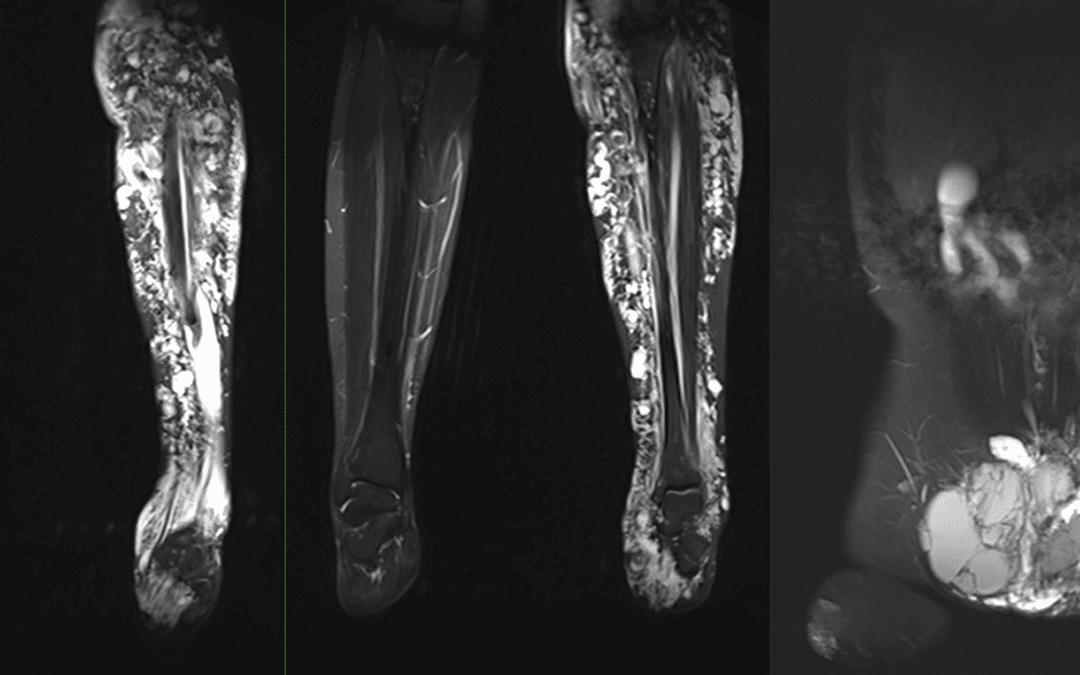
Sehr häufig findet sich beim Klippel-Trénaunay-Syndrom eine Kombination aus kapillären Malformationen, sowie venösen und lymphatischen Malformationen und ein Überwuchs der betroffenen Extremität. Genetisch liegt ein Mosaik vor, das bedeutet, dass Zellen in erkrankten und gesunden Abschnitten genetisch unterschiedlich aufgebaut sind. Aufgrund der Seltenheit und Komplexität dieses Syndroms kommt es gelegentlich zu Unsicherheiten der Nomenklatur.
Die führenden Symptome sind übermäßige Größe und Länge der betroffenen Gliedmaße sowie eine vermehrte Kapillardurchblutung im Bereich der kapillären Malformationen sowie in den gegebenenfalls stark erweiterten Venen. Die erhöhte Kapillardurchblutung führt in der Magnetresonanzangiografie aufgrund der raschen Füllung der Venen gelegentlich zur Fehldiagnose einer AV-Malformation.
Das erhöhte venöse Blutvolumen führt zu häufigen Thrombosen und Venenentzündungen und macht eine langzeitige Kompression mittels Strumpf erforderlich. Invasive Therapien richten sich nach dem Beschwerdebild und sind höchst individuell an den Patienten anzupassen. In erster Linie kommen dabei ein Sklerotherapie und kombinierte endovenöse Lasertherapie der erkrankten Venensegmente in Frage. Eine Heilung ist bislang nicht möglich.